
Our research-readiness assessments provide an at-a-glance view of the gaps that should be addressed before and during the execution of a drug-discovery project. We also create bespoke reports that provide a detailed analysis of the readiness of your ultra-rare disease of interest, helping you sharpen your research strategy and prioritize your goals. Get in touch to find out how we can help your organization.
FBXO11-related neurodevelopmental disorder. This condition (also known as IDDFBA (intellectual developmental disorder with dysmorphic facies and behavioral abnormalities) results from mutations in the gene coding for F-box only protein 11, a part of the E3 ubiquitin-ligase SCF complex. Our assessment shows that this field is at an early stage of maturity, with multiple gaps across the different domains we evaluate. July 31, 2026
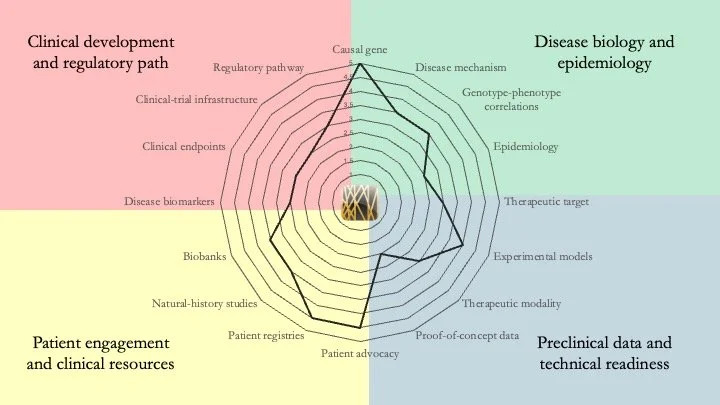
GNAO1-related disorder. This disease is caused by mutations that affect the function of G-protein alpha O1, a brain-enriched protein that seems to have a key role in neurotransmitter release. Patients living with GNAO1-related disorder experience developmental delays, epilepsy, and movement disorders such as dystonia and chorea. Our assessment shows that a lot of mechanistic work remains to be done, but a recent study by L. R. Heideman et al. (Dev. Med. Child Neurol. Feb. 2026) defining the assessment tools that should be used in a GNAO1 registry is an important step in the right direction for this field. July 15, 2026
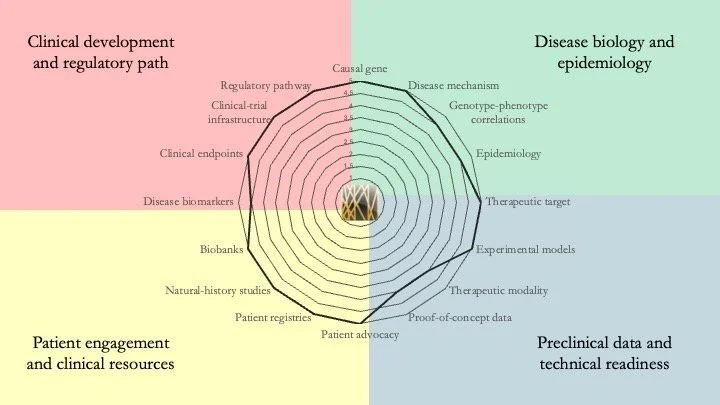
Fibrodysplasia ossificans progressiva. FOP is caused by mutations in the ACVR1 gene, which encodes ALK2, a receptor involved in the BMP signaling pathway. In this disease muscles, ligaments and tendons progressively turn into bone. Although palovarotene has been approved to treat FOP, a definitive cure has yet to be found. However, our assessment shows that its research readiness is essentially complete, and new disease-modifying therapies (saracatinib, garetosmab) are in late-stage clinical development. July 9, 2026.
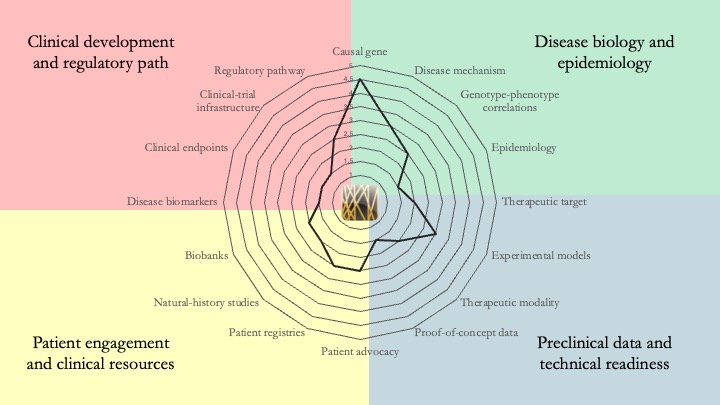
CASK-related neurodevelopmental disorder. This is an X-linked genetic condition that presents in two main forms: a severe form with progressive microcephaly and brain underdevelopment, and a milder form featuring intellectual disability with or without nystagmus. Our research-readiness assessment shows that patient engagement and clinical resources are ahead of the curve for an ultra-rare condition. The field has also taken important strides in disease biology and model development, and is making steady progress on the early translational work required before clinical development. July 2, 2026
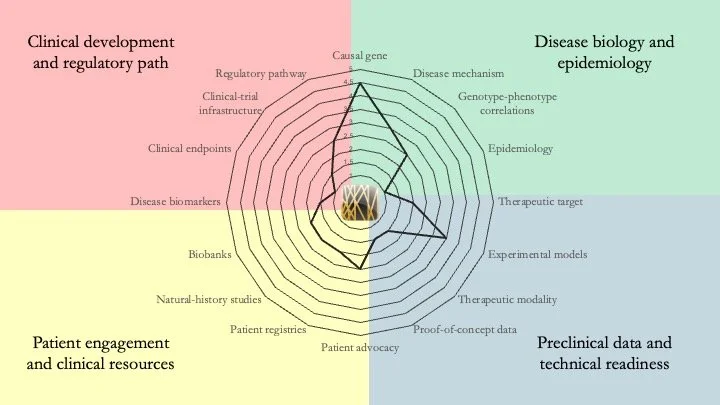
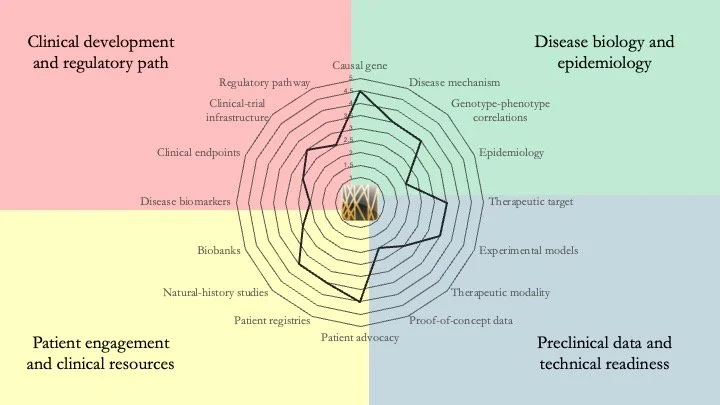
MAPK8IP3-related neurodevelopmental disorder. This condition results from mutations in the gene encoding JIP3 (JNK-interacting protein 3), which links cargo to dynein and kinesin, and is involved in axonal transport and guidance. Although the mechanistic understanding of the disease is somewhat understood, a lot of work remains to be done, particularly in terms of technical readiness and clinical resources. June 24, 2026.
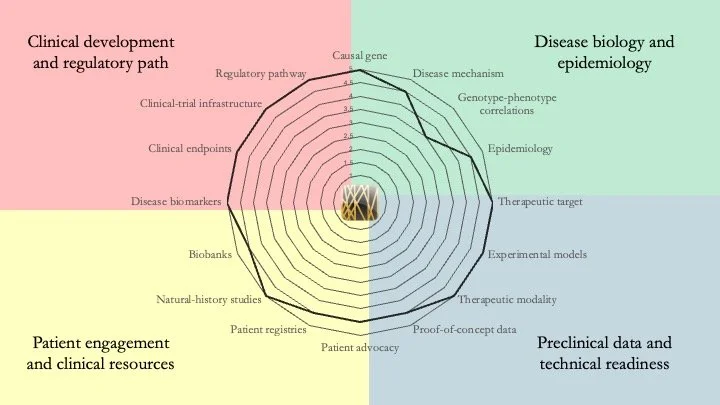
ATP7B-related Wilson disease. This autosomal recessive disorder results from mutations in the gene encoding a copper-transporting protein ATPase. This leads to a failure to excrete copper, leading to its toxic accumulation in the multiple organs. This condition a long history of clinical development and several approved therapies (copper chelators). Current work in the field has focused on the development of advanced therapeutics to improve efficacy and durability. This disease represents a near-ceiling example of research readiness. June 18, 2026
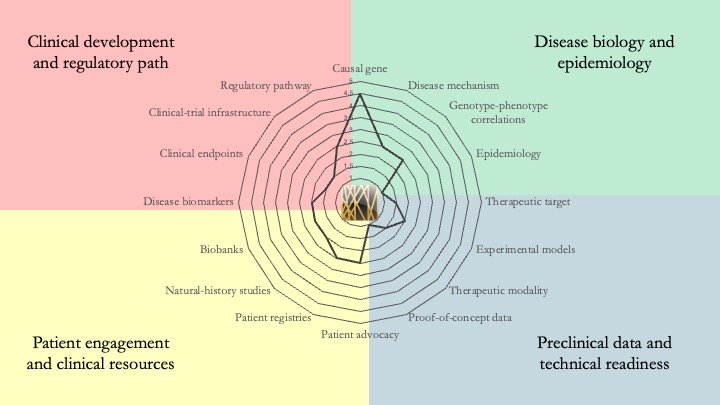
HNRNPK-related neurodevelopmental disorder. Also known as Au-Kline Syndrome, this condition is caused by mutations in the gene that encodes hnRNP K, an RNA-binding protein that regulates gene expression and is important during brain development. Our understanding of hnRNP K is extensive, but most of what we know comes from its overexpression in cancer and its role in erythroid differentiation. A key priority for this disease is the generation of new models (such as iPSCs from patients) to interrogate hnRNP K biology in the nervous system. June 10, 2026
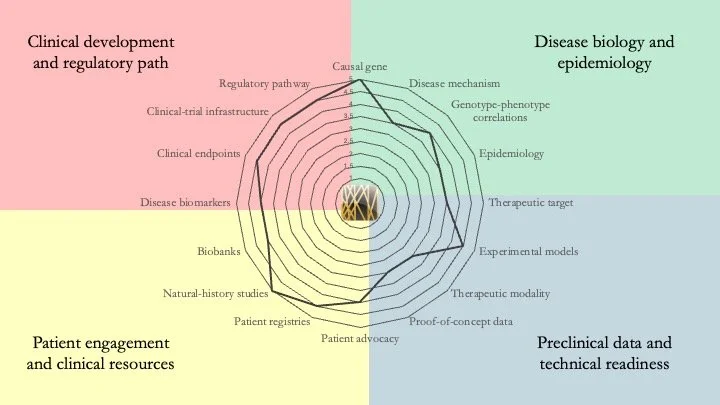
LMNA-related dilated cardiomyopathy. This disease is caused by mutations in the LMNA gene. It typically presents in early to mid-adulthood, featuring an enlarged left ventricle and severe alterations of electrical conduction. It scores very high in our research-readiness assessment; the biological understanding of the condition and the clinical resources available provide a clear roadmap for the development of a therapy. In fact, Pfizer already conducted a (failed) Phase 3 trial of a p38 MAPK inhibitor. New therapeutic approaches include gene therapy and allele-specific shRNA silencing of the mutant LMNA allele. June 3, 2026.
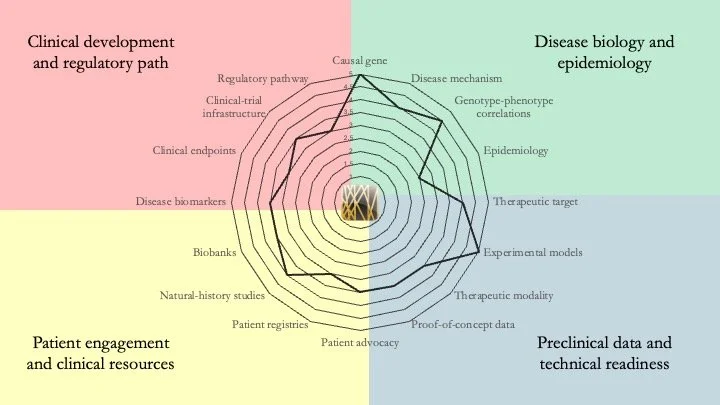
Pelizaeus–Merzbacher Disease. PMD is an X-linked leukodystrophy caused by mutations in the PLP1 gene. Patients typically experience hypotonia, nystagmus, spasticity, ataxia and dystonia. Our research-readiness assessment indicates that understanding of the disease is rather mature across all dimensions, with a couple of ASO- and stem cell-based clinical programs already taking place. However, a cure remains elusive. May 27, 2026.
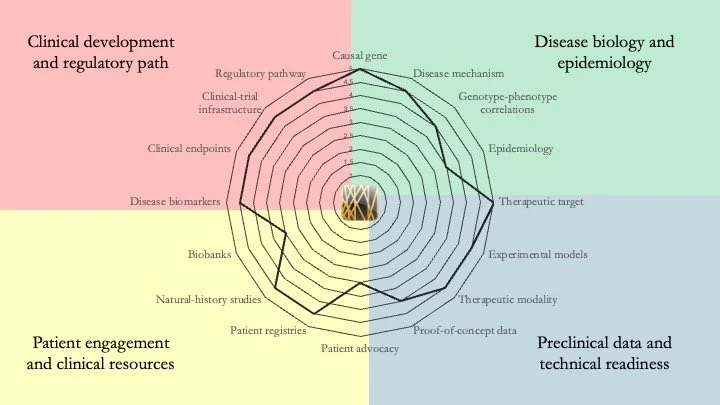
Lysosomal acid lipase deficiency. This is an autosomal recessive disease caused by mutations in the LIPA gene. Manifestations of the disease include accumulation of cholesteryl esters and triglycerides in liver, spleen and other organs. If left untreated, it can be fatal, particularly in pediatric patients. Our research-readiness assessment shows that patients with this condition could benefit from gene therapy. However, as enzyme-replacement therapy is available, there is little incentive to develop a curative therapy. This is one case in which patient-advocacy groups can make a difference by picking up the slack that commercial drug developers are not interested in. May 20, 2026.
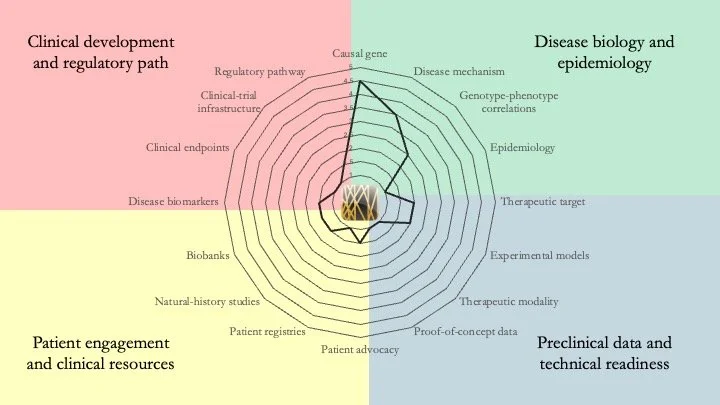
CHOPS Syndrome. This condition, discovered in 2015 and with <50 known cases worldwide, is caused by heterozygous, gain-of-function mutations in AFF4. It is a multi-system disorder characterized by cognitive impairment, heart and lung defects, obesity and skeletal abnormalities. Although the mechanistic understanding of the disease is limited, PROTACs or molecular glues could be suitable therapeutic modalities. However, the path to a therapy remains long. May 14, 2026.
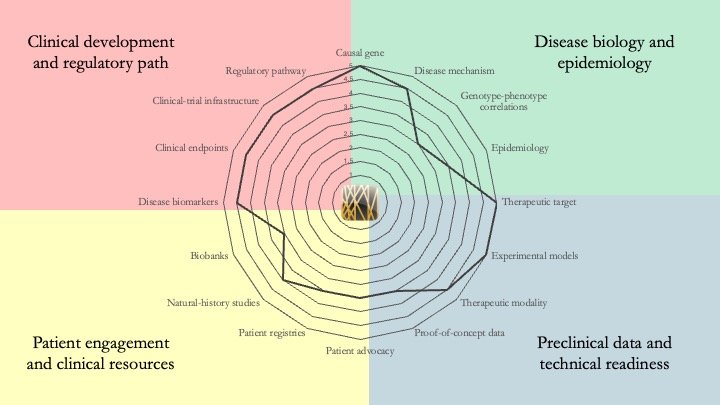
Alpha-mannosidosis. Although an enzyme-replacement therapy has been approved for this condition, it does not address the neurological effects of this multi-system disease. Future therapies must tackle this limitation. In terms of research readiness, genotype-phenotype correlations remain an important gap for alpha-mannosidosis but, by contrast to most ultra-rare diseases, it scores very high on clinical readiness. May 11, 2026.
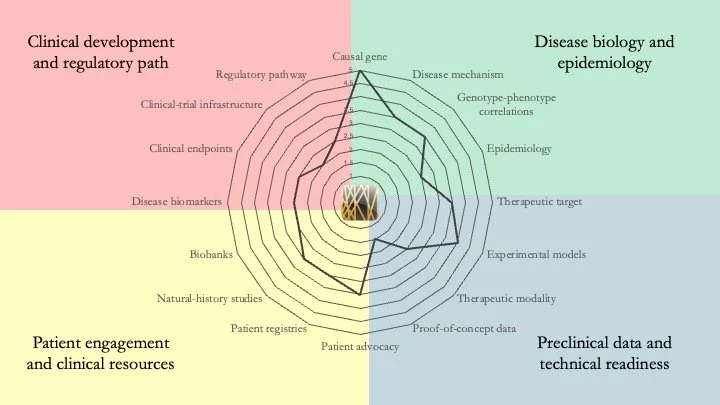
KAT6A Syndrome. Also known as Arboleda-Tham Syndrome, this neurodevelopmental condition results from mutations in the gene encoding lysine acetyltransferase 6A. Our research-readiness assessment indicates that the molecular mechanism underlying the pathology is increasingly understood, as exemplified by recent studies by Liu et al. (Science Advances) and Bergamasco et al. (Nature Communications). However, significant gaps remain in technical and clinical readiness. May 8, 2026.
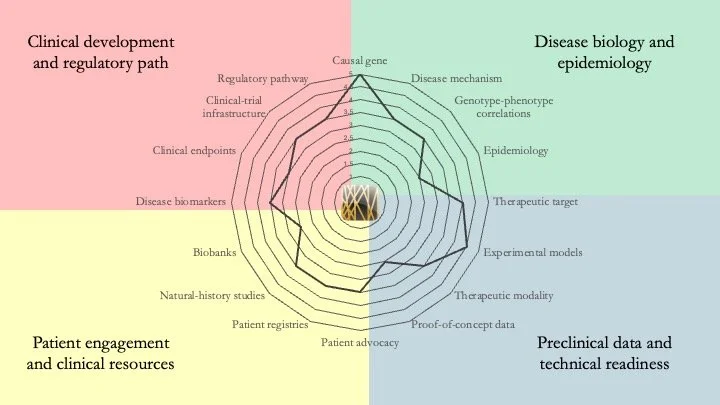
ADNP Syndrome. Also known as Helsmoortel–Van der Aa Syndrome, this is a neurodevelopmental disorder caused by mutations in the gene encoding Activity-Dependent Neuroprotective Protein. Our assessment shows that this condition is sufficiently mature for clinical development. In fact, clinical programs have already been launched. However, gaps remain in terms of mechanistic understanding and clinical resources. April 30, 2026.
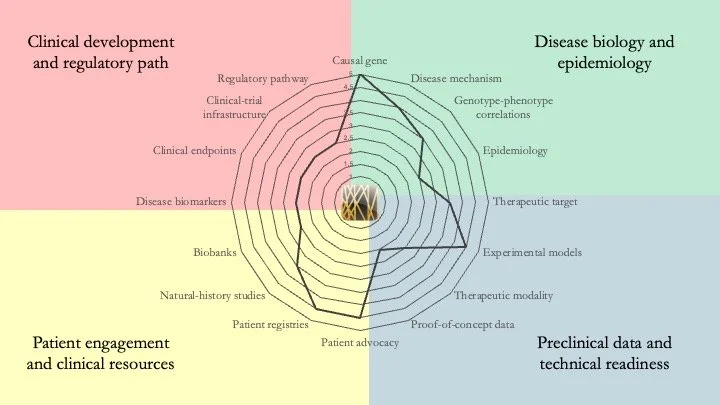
DYRK1A Syndrome. DYRK1A haploinsufficiency results in developmental delay, intellectual disability and microcephaly. There is a lot of preclinical work around this condition, and a recent study by Ron et al. in the journal Molecular Psychiatry on the therapeutic effect of lithium on a mouse model stands out as particularly important. April 27, 2026.
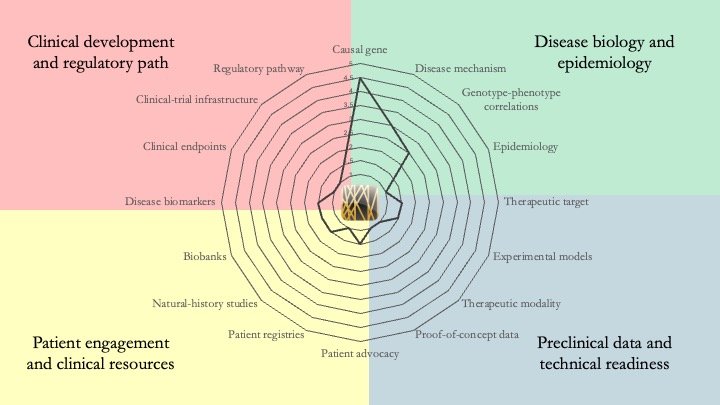
TFE3-associated neurodevelopmental disorder. Only a few dozen patients have been identified since the disease was first described in 2019, which explains why multiple readiness gaps remain to be addressed. But what makes the condition particularly interesting is that the underlying mechanism may be amenable to the use of molecular glues, something that isn't always the case for genetic diseases. April 23, 2026.
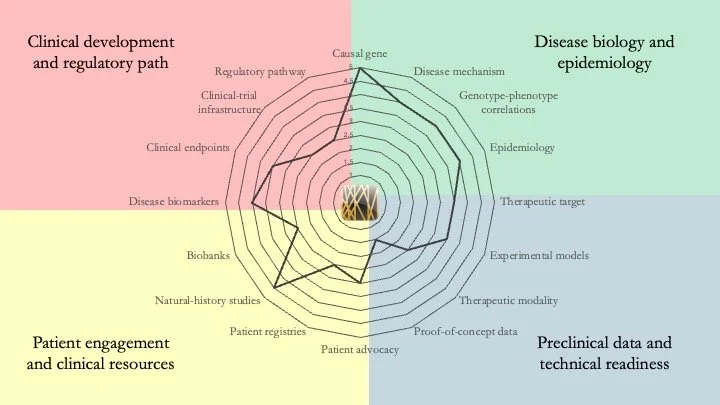
HRPT1 deficiency. HRPT1 encodes hypoxanthine-guanine phosphoribosyltransferase, a key enzyme in the purine salvage pathway. Its deficiency results in a variety of diseases, some of which are very severe (e.g. Lesch-Nyhan Disease). Our assessment indicates that phenotype-genotype correlations are well established for this group of diseases, but the absence of robust animal models has prevented translational progress. April 16, 2026.
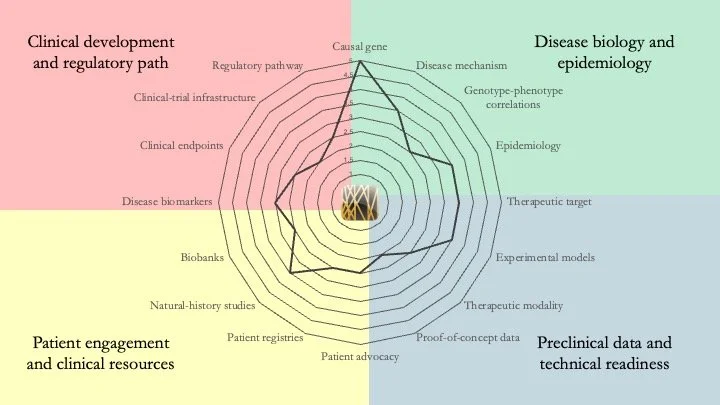
Glycogen Storage Disease IV. Also known as Andersen Disease, this condition is cause by mutations in GBE1, which encodes 1,4-alpha-glucan branching enzyme 1. Broadly speaking, there are two forms of the disease: a pediatric form and an adult-onset form. The research readiness of the two forms is different, and the above plot shows a composite score, which discloses multiple gaps in different domains. April 13, 2026.
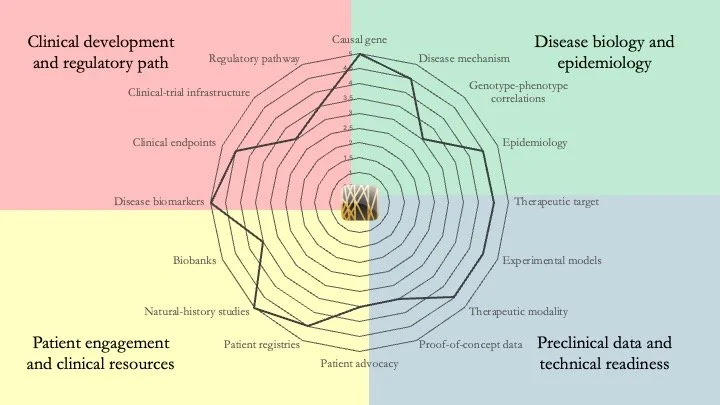
Glutaric Aciduria Type 1. This disease is an inborn error of metabolism caused by mutations in the GCDH gene, preventing the breakdown of lysine, hydroxylysine and tryptophan. It is accompanied by metabolic crises and neurological damage. Fortunately, our understanding of the condition is quite mature and on the cusp of clinical translation. April 9, 2026.
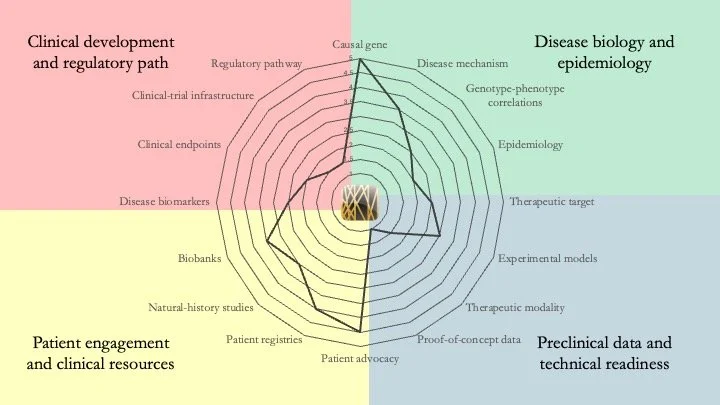
Bohring-Opitz Syndrome. This is a multi-system disorder caused by mutations in ASXL1, which codes for a protein involved in chromatin remodeling. Our analysis shows that a lot of work remains to be done in terms of technical readiness and laying the foundation for eventual clinical development. An immediate priority would be to identify the right therapeutic modality to use and the right phenotype to target. April 6, 2026.
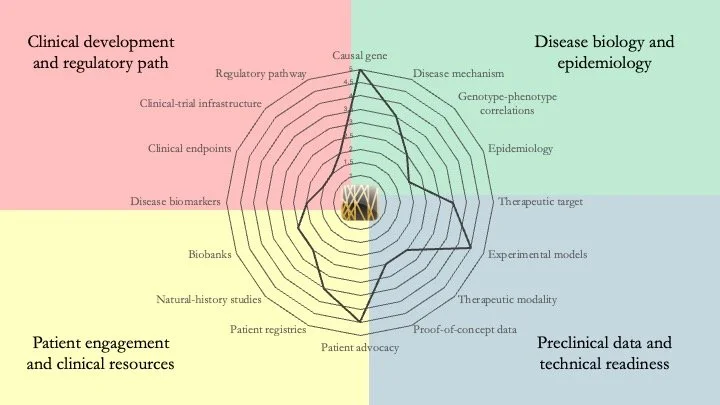
MEF2C Haploinsufficiency Syndrome. This condition, caused by mutations in one copy of the transcription factor MEF2C, results in severe developmental delay, epilepsy and sleep disturbances, among other symptoms. Our research-readiness assessment shows that, although the disease mechanism is increasingly well understood, key gaps include the paucity of rescue data in preclinical models using any disease-modifying therapeutic modality and the absence of clinical-trial infrastructure. April 2, 2026.
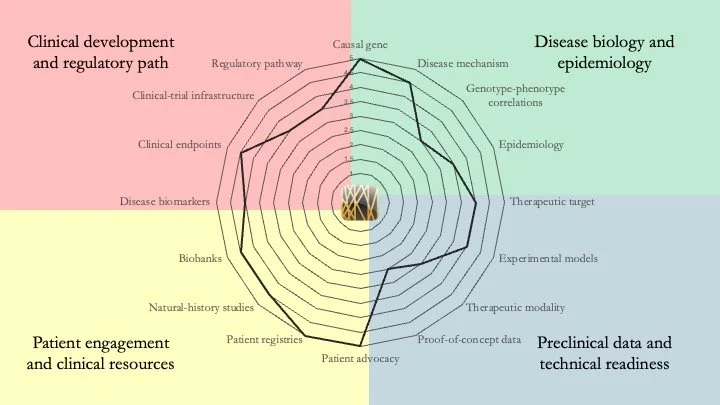
Shwachman-Diamond Syndrome. This multi-system disorder is caused by mutations in the SBDS gene, a protein important for ribosomal biogenesis. The disease is characterized by bone-marrow failure, pancreatic insufficiency, skeletal abnormalities and other phenotypes. The main gaps that we identified pertain to the lack of in vivo data showing phenotypic rescue after therapeutic interventions. The emergence of new experimental models may help address this issue. March 30, 2026.
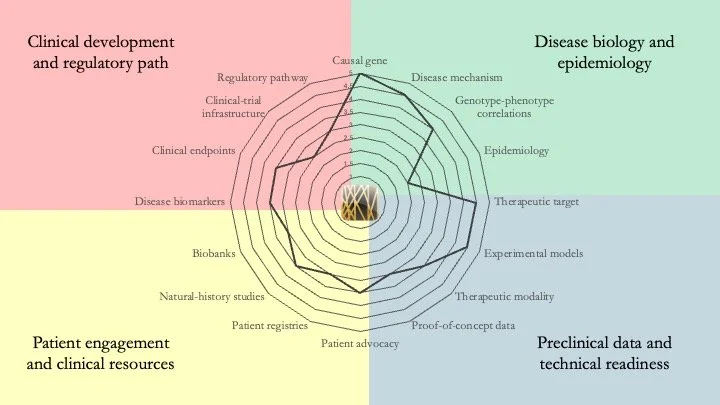
Timothy Syndrome. This channelopathy is characterized by the presence of arrhythmias and neurodevelopmental phenotypes. Timothy Syndrome is caused by gain-of-function mutations in CACNA1C, which encodes the voltage-gated calcium channel Cav1.2. Our analysis shows that proof-of-concept data remain scarce, with ASOs being the preferred modality so far. Key gaps also remain in terms of clinical readiness and establishing the prevalence of the syndrome. March 26, 2026.
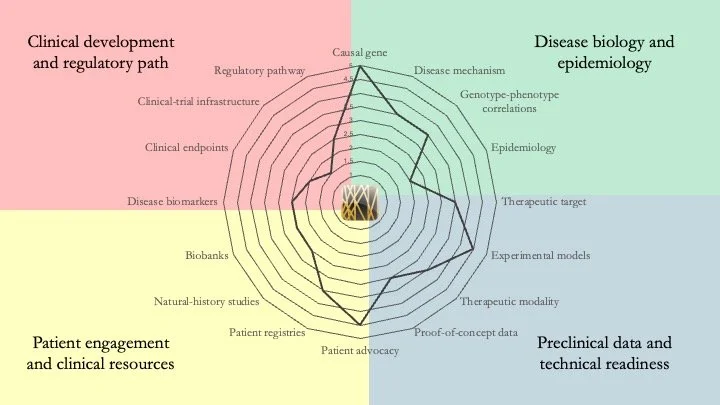
PACS1-related syndrome. Also known as Schuurs-Hoeijmakers Syndrome, this neurodevelopmental disorder is caused by mutations in a protein involved in trafficking through the Golgi network. The existence of strong genotype-phenotype correlations point to ASOs as the ideal therapeutic modality for this disease. However, important gaps remain on the clinical-development side. March 23, 2026.
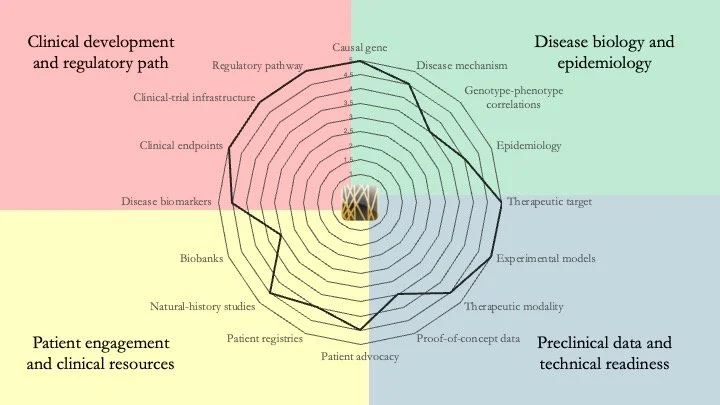
Gorlin Syndrome. Condition caused by mutations in PTCH1 and characterized by the development of basal cell carcinomas and other tumors, as well as skeletal abnormalities. Our analysis shows very few gaps, which is not unexpected, considering the availability of therapies for this condition. But as the existing therapies are not curative, Gorlin Syndrome may be fertile ground for the development of advanced therapeutics. March 19, 2026.
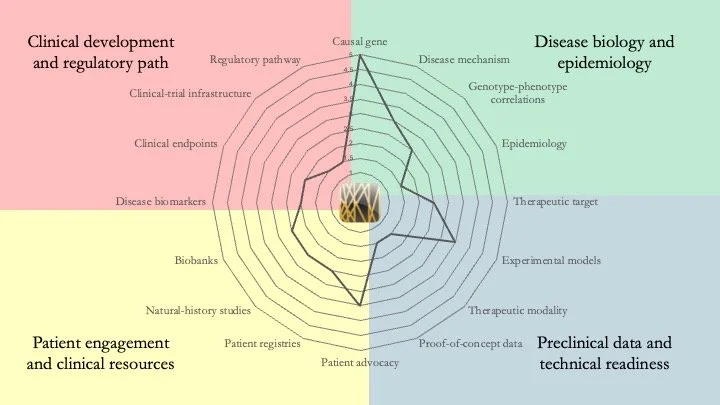
ADSSL1 Myopathy. Mutations in the ADSSL1 gene, which encodes adenylosuccinate synthetase 1, affect purine metabolism and energy production in muscle cells. Our analysis shows that multiple gaps remain across the different domains that we evaluate, starting with a limited understanding of disease biology. March 16, 2026.
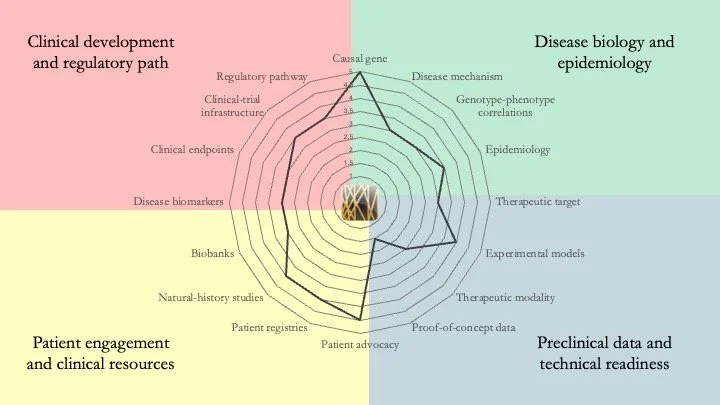
Alström Syndrome. Multi-system disorder caused by mutations in the ALMS1 gene. Our analysis indicates that important gaps remain in understanding disease mechanisms and in the identification of the ideal modality to develop a therapeutic. March 12, 2026.